Each person’s DNA comprises about six billion letters of code. Less than 1% of this code differs from one person to the next. These small differences contribute to each person’s uniqueness and can provide insights about their health and diseases.
Whenever a new genome is sequenced, scientists rely on the reference human genome as a guiding template. The reference human genome was an outcome of the Human Genome Project that ended 20 years ago. In the early 2000s, sequencing DNA was orders of magnitude more expensive and laborious compared to today. As a result, the original reference sequence was constructed by sequencing the genetic material of only a few people. Unfortunately, this led to an inadequate representation of human diversity in the reference sequence.
The research community has been working to address this problem. In 2023, an NIH-funded consortium published the first human pangenome reference. The term ‘pangenome’ implies a collection of genome sequences derived from many individuals. For example, this new published human pangenome reference comprised genomes of 47 people from diverse populations. Anyone who downloads the original older reference human genome sequence would see a document containing strings of A, C, G and T characters. This representation works fine for saving the genome sequence of a single person. However, the same string-based representation cannot be used for a pangenome reference because it needs to also reflect the genomic differences across multiple individuals. Accordingly, the human pangenome reference is represented as a network. It is also referred to as “pangenome graph”. The shared sequences among the sequenced individuals are represented as common nodes while the genomic differences appear as bubble-like structures in this network.
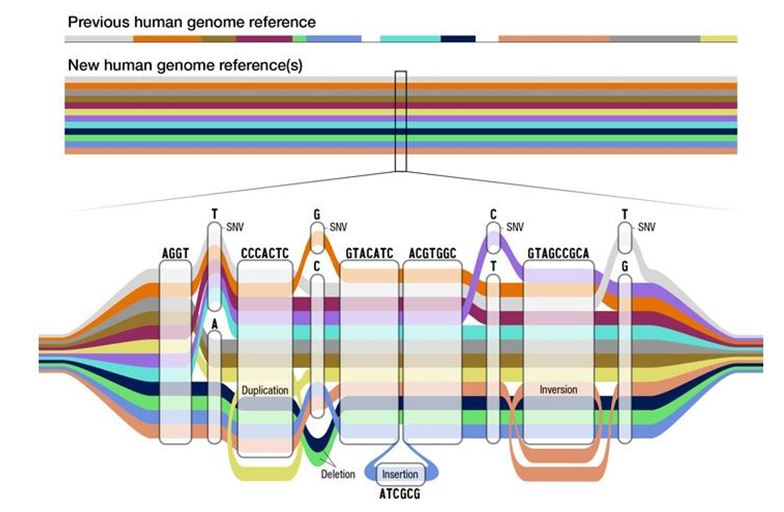
Using this new pangenome reference for the standard genomic applications is not straightforward. For example, sequencing a genome of a person now requires finding a matching route for their sequences through the graph. This task is referred to as sequence- to- graph alignment in bioinformatics.
A new collaborative study between the Indian Institute of Science (IISc) and the University of Texas Dallas has now provided novel ways for mathematically formulating the sequence- to-graph alignment problem and solving them using fast computer algorithms. This study, funded by IndiaAlliance DBT/Wellcome Trust, has been published in Genome Research. The project was led by Ghanshyam Chandra, fifth year PhD student at the Department of Computational and Data Sciences in IISc. Chandra carried out this research under the mentorship of Daniel Gibney and Chirag Jain. The work was also presented this year at the RECOMB 2024 conference held at MIT.
The basic intuition in this study is that a pangenome graph reference not just reflects the biologically plausible genome sequences, but it also reflects a large number of flawed sequences via false recombinations in the graph. The flawed sequences should not be used in downstream applications. The authors provide a solution to overcome this issue by introducing a penalty per recombination in the objective function so that false recombinations can be avoided during sequence alignment. The work employs conventional computer science techniques, including dynamic programming and fine-grained reductions. The proposed algorithms are available as open-source software on GitHub. Through this project, scientists now have access to superior algorithms for genome resequencing in basic and clinical biomedical applications.
REFERENCE:
Chandra G, Gibney D, Jain C, Haplotype-aware sequence alignment to pangenome graphs, Genome Research (2024).
https://genome.cshlp.org/content/early/2024/07/16/gr.279143.124
Link to open-access preprint: https://www.biorxiv.org/content/10.1101/2023.11.15.566493v2
LAB WEBSITE:
https://at-cg.github.io/